
Following closely on the landmark agreement between the European Parliament and the Council on the EU’s “pharma package”, regulatory change is now extending into the realm of medical device law.
On 16 December 2025, the European Commission published a proposal to simplify Regulation (EU) 2017/745 on medical devices (the “MDR”) and Regulation (EU) 2017/746 on in vitro diagnostic medical devices (the “IVDR”), COM(2025) 1023 final, available here.
The proposal contains numerous and in some cases fundamental amendments aimed at reducing administrative burden, eliminating unnecessary regulatory provisions and improving coordination within the EU. It also proposes important changes to promote innovation and digitalisation.
The key aspects of the planned reform are outlined below.
Key points
1. AI-based medical devices
The European Commission envisages a fundamental realignment between Regulation (EU) 2024/1689 on artificial intelligence (the “AI Act”) and medical device regulations, with the AI Act only to apply to medical devices and IVDs to a very limited extent in future.
The MDR and IVDR would be moved from Section A of Annex I of the AI Act to Section B of Annex I. Since Article 2(2) AI Act largely exempts high-risk AI systems under Section B from the Act’s scope of application, in particular the substantive requirements of the Act applicable to high-risk AI systems would then cease to apply.
However, the proposal stipulates that the Commission must take the requirements of the AI Act applicable to high-risk AI systems into account when adopting implementing and delegated acts (new Article 5(8) MDR).
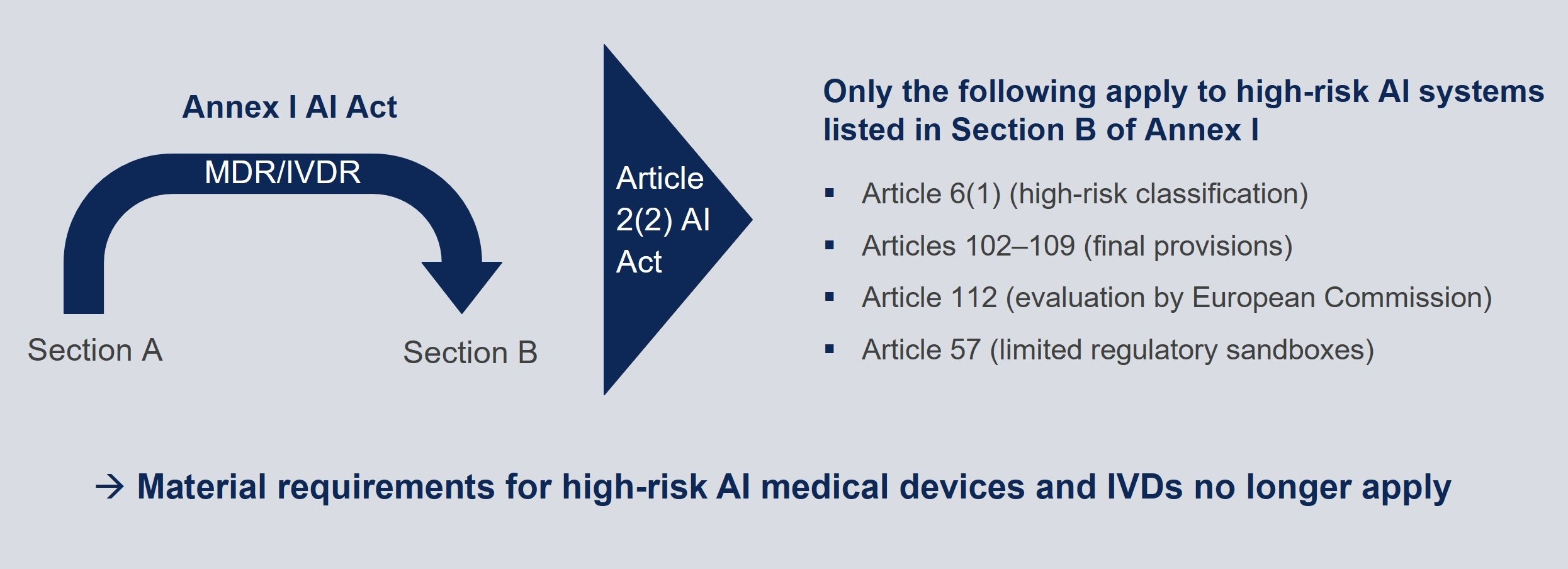
Moreover, notified bodies carrying out conformity assessment for high-risk AI systems pursuant to Article 6(1) AI Act must fulfil certain requirements for notified bodies under the AI Act (Articles 31(4), (5), (10) and (11) AI Act).
Update (7 May 2026) – AI Omnibus: Medical devices and IVDs remain within the full scope of the AI Act. Contrary to the Commission's proposal of December 2025, the Council of the EU and the European Parliament agreed in the so-called AI Omnibus on 7 May 2026 that the MDR and IVDR shall not be moved from Section A to Section B of Annex I – the proposed broad inapplicability of the AI Act to medical devices and IVDs will therefore not be implemented. AI-enabled medical devices and IVDs thus continue to be subject to the full set of high-risk obligations. However, as a compromise, the EU Commission is to be empowered to limit the applicability of the AI Act by way of implementing acts, insofar as the MDR or IVDR ensure an equivalent level of protection. The formal adoption of the AI Omnibus is still pending (source: Press release of the Council of the EU of 7 May 2026).
2. Reduction of administrative burden and elimination of regulatory requirements
2.1 Validity of certificates of conformity (Article 56 MDR, Article 51 IVDR)
The Commission’s proposal specifies that notified bodies may issue certificates of conformity that are valid for an unlimited period, instead of the current maximum period of five years. The notified bodies are however to carry out periodic reviews proportionate to device risk.
2.2 Classification of medical devices (Annex VIII MDR)
The classification rules for medical devices in Annex VIII are to be amended, including the heavily criticised Rule 11 on software classification, which as currently written means that virtually no medical device software is classified as class I.
Under the Commission’s proposal, medical device software that is intended to generate an output that confers a clinical benefit and is used for diagnosis, treatment, prevention, monitoring, prediction, prognosis, compensation or alleviation of a disease or condition will generally be classified as class I. This represents a fundamental change from the current regime, under which software is usually classified as class IIa and class I serves only as a fallback option.
While the subsequent indents still list situations where a higher classification applies, the broad wording could result in software being classified as class IIa simply by virtue of being used in “non-serious situations”. This would exacerbate, instead of solving, the problem of there being hardly any class I software.
2.3 Summary of safety and clinical performance (Article 32 MDR, Article 29 IVDR)
According to the Commission’s proposal, the obligation to draw up a summary of safety and clinical performance (“SSCP”) will no longer apply to “well-established technology devices”.
This obligation would apply to only certain categories of IVDs (companion diagnostics, class C devices for self-testing and class D devices).
A key practical benefit is that the SSCP will no longer have to be validated by the notified body.
2.4 Periodic safety update report (Article 86 MDR, Article 81 IVDR)
Manufacturers will not have to update their periodic safety update reports (“PSUR”) as frequently as before.
The PSUR will only have to be updated every two years – instead of annually – for class IIb and III medical devices and for class C and D IVDs, and only when necessary – instead of every two years – for class IIa medical devices.
Custom-made medical devices are to be exempted from the obligation to submit a safety update report, and this report will therefore no longer be part of the documentation referred to in Section 2 of Annex XIII.
2.5 Deadline for reporting serious incidents (Article 87 MDR, Article 82 IVDR)
The proposal extends the reporting deadline for the specified serious incidents from 15 to 30 days, provided that these incidents are not related to public health threats, death or serious deterioration of health,
3. Promotion of innovation
3.1 Regulatory sandboxes (new Articles 59b and 59c MDR, new Articles 54b and 54c IVDR)
Regulatory sandboxes are to be introduced to promote innovation, enabling (potential) manufacturers to develop, test, validate and use, where appropriate in real-world conditions, innovative products for a limited time under regulatory supervision without having to fulfil all the regulatory requirements.
3.2 In-house medical devices (Article 5(5) MDR, Article 5(5) IVDR)
The proposal introduces more flexibility for the conditions for the manufacture and use of in-house medical devices at health institutions in the EU. It will in future be possible to transfer such devices to other legally independent institutions if this is in the interest of public health. The obligations to provide information to the competent authorities are also to be reduced.
3.3 Breakthrough and orphan devices (new Article 52a MDR, new Article 48a IVDR)
Particularly innovative “breakthrough” devices and orphan devices will be legally defined on the basis of specific criteria. They will be subject to a priority review by notified bodies with the aim of reducing assessment timelines.
In December 2025, the Medical Device Coordination Group also published MDCG 2025-9 “Guidance on Breakthrough Devices (BtX)”, available here.
4. Digitalisation
4.1 Digitalisation of compliance tools (Article 19, new Articles 52b and 110a, Annexes I and VI MDR; Article 17, new Articles 48b and 103a, Annexes I and VI IVDR)
The European Commission moreover plans to digitalise various compliance tools:
- The EU declaration of conformity as well as additional information and documents may be provided in digital form and documentation may be submitted to notified bodies electronically.
- Certain product information (electronic instructions for use for near-patient tests; certain information on the label) may be provided in digital form.
- Economic operators must provide their digital contact in the EUDAMED database.
4.2 Cybersecurity (new Article 87a, Annex I MDR, new Article 82a, Annex I IVDR)
The proposal aims to more effectively embed cybersecurity in medical device law. Actively exploited vulnerabilities or severe incidents as referred to in the Cyber Resilience Act (2024/2847/EU) are in future to be reported to the national computer security incident response teams (CSIRTs) and the European Union Agency for Cybersecurity (ENISA).
Cybersecurity will also be explicitly incorporated into the general safety and performance requirements of Annex I to the MDR and IVDR.
5. Easing of conformity assessment procedure requirements
5.1 Clinical evidence, clinical and non-clinical data (Article 2, point (48), Article 61, Annexes II and XIV MDR, Annex XIII IVDR)
The provisions on clinical evidence and clinical and non-clinical data are to be expanded and the conditions for using clinical data from an equivalent device are to be made more flexible for certain devices. In some cases, it will also be possible to demonstrate a device’s safety and performance based on non-clinical evidence alone. In addition, the use of innovative testing methods such as in vitro, ex vivo and in silico testing, computational modelling or simulation and pre-clinical evaluation will be supported.
5.2 Notified bodies in the conformity assessment procedure (Article 52, Annexes IX, X and XI MDR, Article 48, Annexes IX, X and XI IVDR)
The Commission’s proposal amends the extent to which notified bodies are involved in the conformity assessment procedure and introduces the option of remote audits.
5.3 Combined studies involving medicinal products, medical devices and/or IVDs (new Article 79a MDR, new Article 75a IVDR)
For combined studies involving medicinal products, medical devices and/or IVDs, a single application will in future be sufficient to trigger a coordinated assessment in accordance with Regulation 536/2014/EU on clinical trials.
5.4 Fees charged by notified bodies (Article 50 MDR)
The notified bodies are to apply, in their lists of standard fees, a fee reduction of at least 50% for manufacturers that are micro enterprises, of at least 25% for small enterprises and at least 50% for orphan devices. The European Commission will be authorised to specify the structure and level of fees charged by notified bodies.
6. Improved coordination and cooperation
A range of measures are intended to improve the coordination and cooperation between various authorities and other stakeholders, for example with regard to the status and classification of products (Article 4, new Articles 4a, 51a, 51b MDR, Article 3, new Articles 3a, 47a, 47b IVDR).
The assessment of notified bodies will be streamlined through the involvement of joint assessment teams (Articles 36-44 MDR, Article 31 IVDR). The complete re-assessment of notified bodies, which previously had to be carried out every five years, will be done away with.
In the event of disputes between manufacturers and notified bodies, the authorities of the Member State will mediate (Article 35 MDR, Article 31 IVDR).
The European Medicines Agency (“EMA”) will provide scientific, technical and administrative support for the coordination between the national authorities (Article 106b MDR).
7. Improved availability
Structural challenges arising from supply bottlenecks will be addressed through new information-sharing and cooperation requirements. Member States will have to be informed of possible shortages in their markets at an early stage. The EMA will monitor bottlenecks and provide support to national authorities and the European Commission. In addition, a list of critical medical devices is to be compiled and an IT portal set up for reporting supply interruptions.
Conclusion and outlook
The European Commission’s strategy of simplifying the complex regulatory framework of the MDR and IVDR – which is often seen as overly burdensome in practice – is to be welcomed. The proposed changes would enable medical device manufacturers to access the market quickly without jeopardising the high level of health protection. Reducing regulatory requirements and administrative obligations after the product has been placed on the market is an important step towards easing the compliance burden on the industry.
The planned relief measures for AI-based medical devices are particularly significant. Following considerable efforts to harmonise the MDR/IVDR and the AI Act and to avoid regulatory overlap, the proposal to largely exclude AI-based medical devices from the scope of the AI Act comes as a surprise. From a practical point of view, this step is a positive development, as it would significantly ease the burden on manufacturers. Yet, whether this far-reaching reversal by the European Commission will actually be implemented in the proposed form remains to be seen.
The proposed amendment to the MDR and the IVDR has been submitted to the European Parliament and the Council of the European Union. How long the legislative process will take – and how it will unfold – is yet to be determined.
