
Nach jahrelangen Verhandlungen über ein europäisches Regelwerk zur Bewertung von Gesundheitstechnologien (Health Technology Assessment – HTA) ist am 11. Januar 2022 die EU-HTA-Verordnung 2021/2282 in Kraft getreten. Nicht zuletzt unter dem Eindruck der COVID-19-Pandemie verspricht sich die EU von den neuen Vorschriften vor allem einen breiten, zügigen Zugang der PatientInnen zu lebenswichtigen und innovativen Gesundheitstechnologien. Gleichzeitig setzt sie auf Effizienzgewinne für die Unternehmen und die Mitgliedstaaten. Im Fokus stehen dabei die gemeinsamen klinischen Bewertungen durch die sog. Koordinierungsgruppe, die für Medizinprodukte und In-vitro-Diagnostika frühestens ab Januar 2025 durchgeführt werden. Trotz des dreijährigen Aufschubs lohnt sich bereits jetzt ein Blick auf den wesentlichen Inhalt der HTA-Verordnung und deren Integration in das deutsche Medizinprodukte- und Sozialversicherungsrecht.
Das Wichtigste zusammengefasst:
I. Gesundheitstechnologien und ihre Bewertung
Als Health Technology Assessment bezeichnet man – vereinfacht gesagt – ein Verfahren, bei dem auf systematische und transparente Weise Informationen zu einer bestimmten Gesundheitstechnologie, ihrer Funktionsweise und ihren Auswirkungen auf ein Gesundheitssystem gesammelt, geordnet, analysiert und bewertet werden. Dabei ist der Begriff der Gesundheitstechnologie umfassend zu verstehen: Gemeint sind nicht nur Arzneimittel, Medizinprodukte und In-vitro-Diagnostika, sondern etwa auch chirurgische Verfahren, Präventionsmaßnahmen oder Diagnose- und Behandlungsmethoden. Typischerweise beleuchtet ein HTA
- medizinische bzw. klinische (v.a. relative Wirksamkeit und relative Sicherheit im Vergleich zu anderen Technologien),
- gesundheitsökonomische (v.a. Kosten und Einsparungen im Vergleich zu anderen Technologien),
- rechtliche (bspw. Zulassung; Haftung; Patientenautonomie; Datenschutz),
- soziale (bspw. Zugangshindernisse für bestimmte Bevölkerungsgruppen),
- ethische (bspw. Fragen der Stigmatisierung; Priorisierung von Ressourcen)
- und/oder organisatorische Aspekte (bspw. Einbindung der Technologie in bestehende Strukturen; Ausbildung des Gesundheitspersonals). Diese Themenfelder, mit Ausnahme der medizinischen Aspekte, firmieren auch als „nichtklinische Bewertung“.
Am Ende des Prozesses steht ein HTA-Bericht, der in erster Linie gesundheitspolitische Maßnahmen vorbereiten und die zuständigen Gremien eines Gesundheitssystems bei der Entscheidung unterstützen soll, ob und zu welchen Konditionen eine Gesundheitstechnologie in die medizinische Regelversorgung aufzunehmen ist.
II. Neuer, gemeinschaftlicher Rahmen für HTA-Verfahren in der EU
Bislang werden diese HTA-Verfahren auf nationaler Ebene in Auftrag gegeben und durchgeführt, in Deutschland etwa durch den Gemeinsamen Bundesausschuss (G-BA) und das Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen (IQWiG). Der Zuschnitt der HTA-Verfahren und die Anforderungen an die wissenschaftlichen Nachweise, die Entwickler von Gesundheitstechnologien für das HTA vorzulegen haben, können in den Mitgliedstaaten jedoch stark voneinander abweichen. Die Industrie sieht sich also mit der Schwierigkeit konfrontiert, das Design einer Studie – bei Arzneimitteln schon auf Ebene der Zulassung – möglichst erfolgversprechend anzulegen.
Um Doppelarbeit auf Seiten der Unternehmen und der Mitgliedstaaten zu vermeiden, schafft die Verordnung (EU) 2021/2282 nun ein neues, EU-finanziertes Grundgerüst für ein HTA-Verfahren auf europäischer Ebene. Die freiwillige und projektbasierte Kooperation der vergangenen Jahrzehnte erfährt damit eine formale Aufwertung. Wesentliche Elemente der HTA-Verordnung sind
- die Einrichtung einer Koordinierungsgruppe sowie eines Netzwerkes der Interessenträger,
- die Durchführung gemeinsamer klinischer Bewertungen und gemeinsamer wissenschaftlicher Beratungen
- sowie die Förderung darüberhinausgehender freiwilliger Zusammenarbeit.
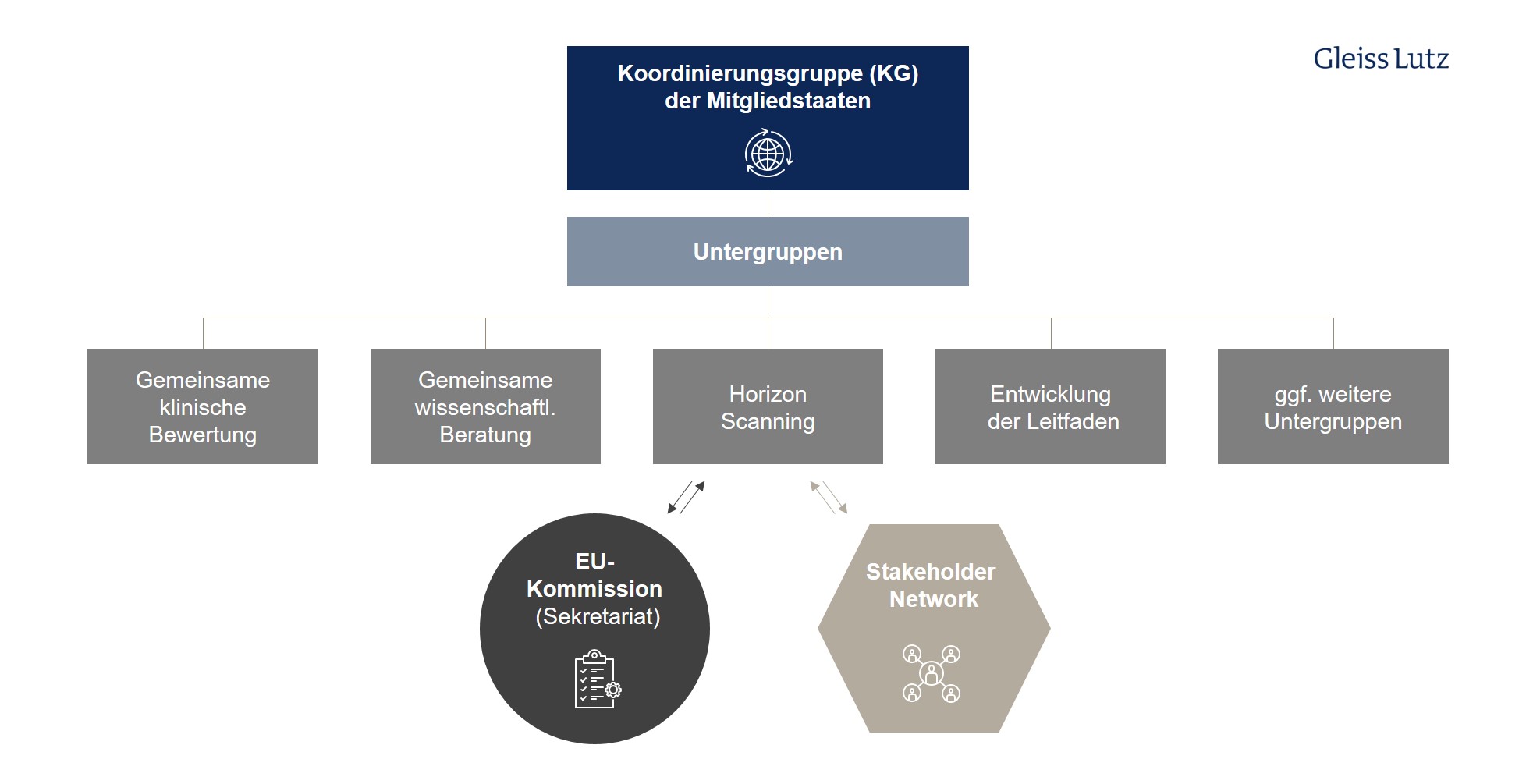
1. Einrichtung einer Koordinierungsgruppe (Art. 3)
Zentraler Akteur der HTA-Verordnung ist neben der Kommission die sog. Koordinierungsgruppe gemäß Art. 3. Ihre Mitglieder, regelmäßig nationale HTA-Stellen wie G-BA und IQWiG, werden von den Mitgliedstaaten benannt.
- Zu den Aufgaben der Koordinierungsgruppe und ihrer Untergruppen gehören die Durchführung der gemeinsamen klinischen Bewertungen und gemeinsamen wissenschaftlichen Beratungen, die Ermittlung neu entstehender Gesundheitstechnologien (sog. Horizon Scanning) sowie die Unterstützung der freiwilligen Zusammenarbeit der Mitgliedstaaten.
- Der Koordinierungsgruppe obliegt außerdem die Entwicklung und Annahme der methodischen Leitfäden, Verfahrensschritte und Fristen, auf deren Grundlage die gemeinsamen Bewertungen und Beratungen durchgeführt werden (Art. 3 Abs 7 lit. d-g).
- Die Verordnung selbst enthält diesbezüglich nur rudimentäre Vorgaben. Für diesen Schritt bleibt der Koordinierungsgruppe nun eine Übergangsphase von drei Jahren; gemäß Art. 36 Abs. 2 gilt die Verordnung erst ab dem 12. Januar 2025.
Die Koordinierungsgruppe wird bei ihrer Tätigkeit von der Kommission unterstützt, die als Sekretariat fungiert. Die ersten Sitzungen sollen bereits im Juni 2022 stattfinden. Hier finden Sie eine graphische Übersicht über die verschiedenen Aufgaben und Untergruppen der HTA Koordinierungsgruppe auf EU-Ebene.
{kind=link}
2. Gemeinsame klinische Bewertungen (Art. 7-15)
Herzstück der HTA-Verordnung ist die gemeinsame klinische Bewertung bestimmter Gesundheitstechnologien (Joint Clinical Assessment – JCA). Die „klinische“ Bewertung beschränkt sich auf
- die Beschreibung des Gesundheitsproblems und die Ermittlung/Beschreibung bestehender Gesundheitstechnologie,
- die Prüfung der technischen Eigenschaften der zu bewertenden Gesundheitstechnologie,
- die Prüfung der relativen Sicherheit
- und die Prüfung der relativen klinischen Wirksamkeit.
Das heißt, dass andere, nichtklinische Aspekte – insbesondere die gesundheitsökomischen Fragen und ein etwaiger Zusatznutzen im Zusammenhang mit Preisbildung und Erstattung – weiterhin von nationalen Stellen analysiert und bewertet werden.
Im Übrigen unterliegen dem EU-HTA zum einen die im zentralen Zulassungsverfahren zugelassenen Arzneimittel nach einem abgestuften Fristenmodell (Art. 7 Abs. 1 lit. a, b, Abs. 2), beginnend mit Arzneimitteln mit einer onkologischen Indikation und Arzneimitteln für neuartige Therapien (2025). Später folgen sog. Orphan Drugs (2028) sowie alle übrigen Arzneimittel (2030).
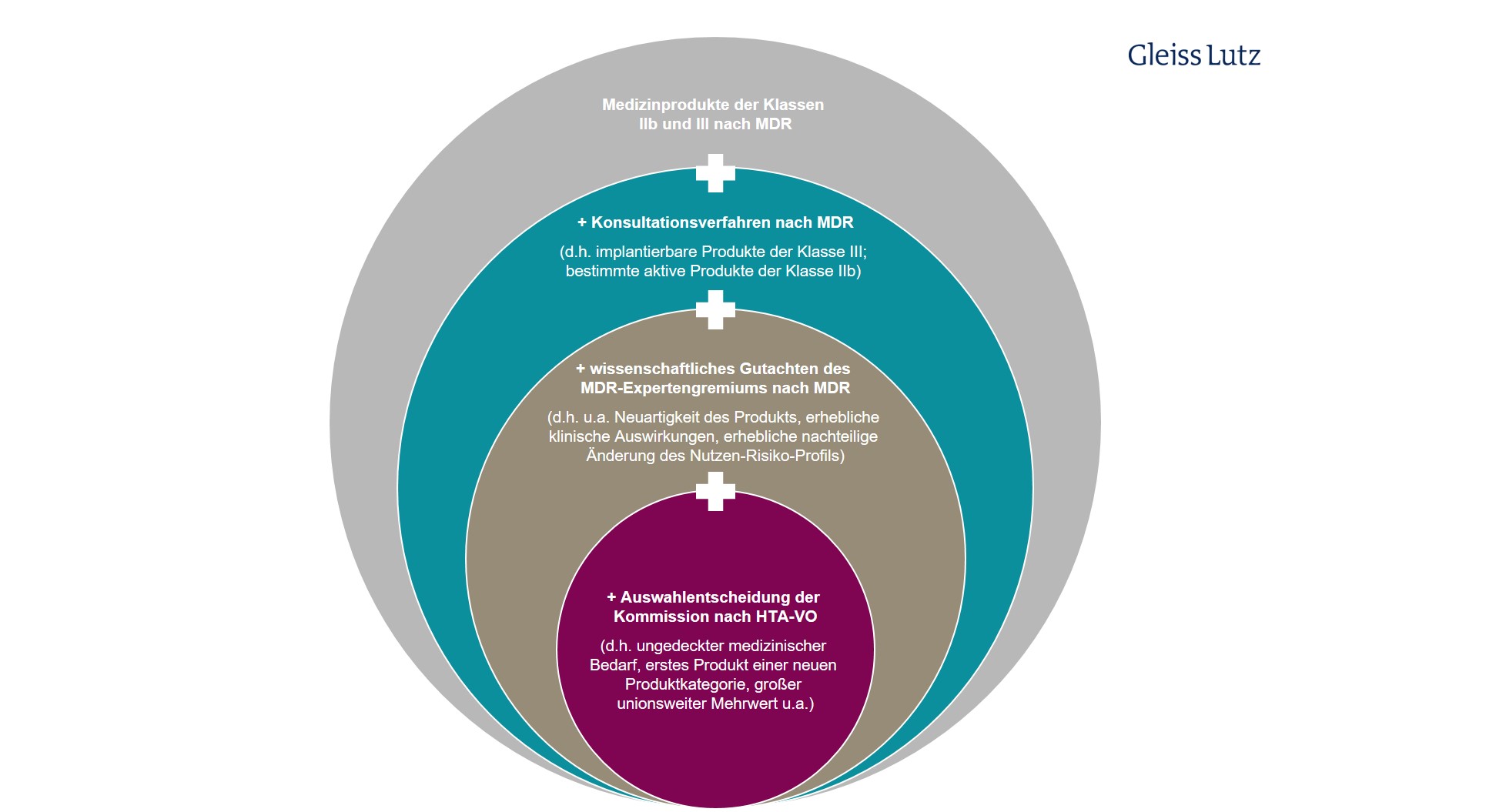
Bei den Medizinprodukten und In-vitro-Diagnostika ist der Anwendungsbereich in inhaltlicher Sicht durch verschiedene Filter eingeschränkt (Art. 7 Abs. 1 lit. c, d). Nur besonders risikoreiche, neuartige und/oder vielversprechende implantierbare Medizinprodukte der Klasse III bzw. aktive Produkte der Klasse IIb im Sinne der Verordnung 2017/745 über Medizinprodukte (MDR) werden auf europäischer Ebene klinisch bewertet.
{kind=link}
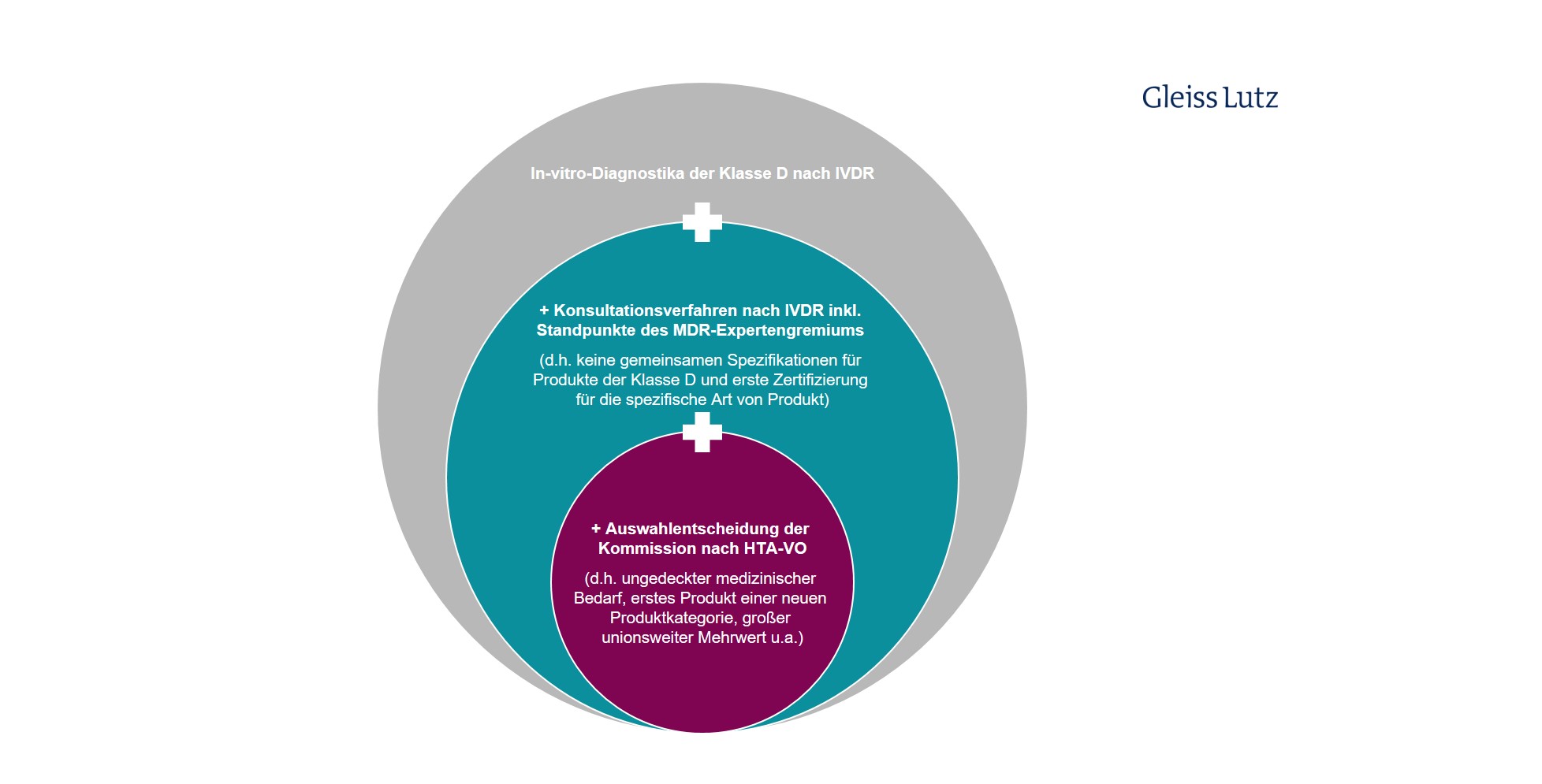
Ähnliche Einschränkungen gelten für In-vitro-Diagnostika. Vom EU-HTA erfasst sind nur bestimmte Produkte der Klasse D im Sinne der Verordnung 2017/746 über In-vitro-Diagnostika (IVDR). Hier finden Sie eine graphische Übersicht zum eingeschränkten Anwendungsbereich des HTA-Verfahrens auf spezielle In-vitro-Diagnostika.
{kind=link}
Überblick über die Verfahrensschritte
Die Durchführung der gemeinsamen klinischen Bewertung obliegt jeweils einer Untergruppe der Koordinierungsgruppe, die aus ihrem Kreis einen Gutachter und einen Mitgutachter aus unterschiedlichen Mitgliedstaaten ernennt. Dadurch sollen Fachkompetenz und Unabhängigkeit gesichert werden.
- Das Verfahren beginnt mit der Festlegung des Bewertungsumfanges (Assessment Scope) durch die Untergruppe, wobei auf die Bedürfnisse der Mitgliedstaaten Rücksicht zu nehmen und Patienten, klinische Experten und andere Sachverständige zu beteiligen sind. (Art. 8 Abs. 6).
- Anschließend fordert die Kommission den Entwickler zur Einreichung eines Dossiers auf (Art. 10 Abs. 1), dessen Inhalt im Falle von Medizinprodukten und In-vitro-Diagnostika durch Anhang II der Verordnung und Durchführungsrechtsakte der Kommission näher bestimmt wird. Wichtig ist, dass das Unternehmen auf nationaler Ebene keine Nachweise vorlegen darf, die bereits auf Unionsebene eingereicht wurden (Art. 10 Abs. 3). Umgekehrt dürfen die Mitgliedstaaten diese Nachweise nicht anfordern (Art. 13 Abs. 1 lit. d).
- Sodann erstellen die Gutachter die Entwürfe eines rein analytischen Bewertungsberichts sowie eines zusammenfassenden Berichts (JCA Report; Art. 11 Abs. 1), die sich auf die relativen Effekte der Gesundheitstechnologie und die Aussagesicherheit der relativen Effekte beschränken und keine Werturteile zum gesamten klinischen Zusatznutzen enthalten dürfen (Art. 9 Abs. 1). Bei der Bewertung sind Anmerkungen von Patienten, klinischen Experten und anderen Sachverständigen sowie der übrigen Mitglieder der Untergruppe zu berücksichtigen (Art. 11 Abs. 3 und 4). Dem Unternehmen selbst wird lediglich das Recht eingeräumt, alle rein technischen oder sachlichen Ungenauigkeiten zu melden; Anmerkungen zu den Ergebnissen des Berichtsentwurfs sind allerdings nicht erlaubt (Art. 11 Abs. 5).
- Anschließend wird der Entwurf von der gesamten Koordinierungsgruppe geprüft (Art. 12 Abs. 1), möglichst einvernehmlich gebilligt (Art. 12 Abs. 2) und von der Kommission nach einer verfahrenstechnischen Prüfung (Art. 12 Abs. 3) auf einer IT-Plattform veröffentlicht (Art. 12 Abs. 4).
Zeitplan
Die Fristen für die einzelnen Verfahrensschritte bei der Bewertung von Medizinprodukten und In-vitro-Diagnostika sind weitgehend der Bestimmung durch die Koordinierungsgruppe überlassen. Aussagen über die Gesamtdauer der gemeinsamen klinischen Bewertung sind daher nur bedingt möglich. Nähere Informationen zum Zeitrahmen dürfte die Verfahrensordnung der Koordinierungsgruppe beinhalten.
Der neuralgische Punkt: (Un-)Verbindlichkeit des Bewertungsberichts als Politikum
Anders als noch der Kommissionsentwurf (COM (2018) 51 final) erklärt die HTA-Verordnung den Bericht über die klinische Bewertung der Gesundheitstechnologie nicht für verbindlich. Diese Frage stand im Mittelpunkt des kontroversen und langwierigen Gesetzgebungsverfahrens und veranlasste mehrere Mitgliedstaaten zur Subsidiaritätsrüge, darunter auch Deutschland. Nunmehr haben die nationalen Stellen den Bericht im Rahmen der nationalen HTA lediglich in angemessener Weise zu berücksichtigen (Art. 13 Abs. 1 lit. a). Im Übrigen betont die Verordnung mehrfach, dass die Zuständigkeit der Mitgliedstaaten, ihre eigenen Schlussfolgerungen über den gesamten klinischen Zusatznutzen zu ziehen und eigenverantwortlich Entscheidungen über Preisbildung und Erstattung zu treffen, unberührt bleibt (Art. 13 Abs. 1 lit. a; Erwägungsgründe 14, 26, 28, 31).
Weiterhin zulässig bleiben auch für den allgemeinen nationalen HTA-Prozess erforderliche ergänzende klinische Analysen sowie von vornherein nicht von der HTA-Verordnung erfasste nichtklinische Bewertungen durch die Mitgliedstaaten (vgl. Erwägungsgrund 15). In diesem Rahmen dürfen die Mitgliedstaaten vom Entwickler auch weitere Nachweise anfordern.
3. Gemeinsame wissenschaftliche Beratungen (Art. 16-21)
Für Gesundheitstechnologien, die voraussichtlich Gegenstand gemeinsamer klinischer Bewertungen sind und bei denen sich die klinischen Studien und Prüfungen erst noch in der Planung befinden, können die Unternehmen eine gemeinsame wissenschaftliche Beratung (Joint Scientific Consultation) mit der Koordinierungsgruppe beantragen. Besprochen werden beispielsweise Studiendesigns mit Blick auf die Patientenpopulationen, Interventionen, Komparatoren und gesundheitsbezogenen Endpunkte (Art. 16 Abs. 1; vgl. PICO-Schema). Entwickler von Medizinprodukten können darüber hinaus beantragen, dass die Beratung parallel zu den Beratungen der Expertengremien nach der Verordnung 2017/745 über Medizinprodukte (MDR) stattfindet (Art. 16 Abs. 5, Art. 17 Abs. 2).
4. Freiwillige Zusammenarbeit (Art. 23)
Auch die freiwillige Zusammenarbeit der Mitgliedstaaten bei HTA wird weiterhin gefördert und durch die HTA-Verordnung in einen neuen organisatorischen und finanziellen Rahmen eingebettet. Dies betrifft insbesondere
- nichtklinische Bewertungen von Gesundheitstechnologien unter wirtschaftlichen, rechtlichen, sozialen, ethischen und/oder organisatorischen Aspekten,
- klinische Bewertungen von sonstigen Medizinprodukten und In-vitro-Diagnostika, die nicht bereits dem EU-HTA nach Art 7 unterliegen,
- HTA von Gesundheitstechnologien, die keine Arzneimittel, Medizinprodukte oder In-vitro-Diagnostika darstellen (bspw. chirurgische Verfahren, Maßnahmen zur Prävention oder Diagnose- und Behandlungsverfahren).
5. Netzwerk der Interessenträger (Art. 29)
Möglichkeiten zur Einflussnahme im Bereich des europäischen HTA ergeben sich für die Medizintechnikindustrie im Wesentlichen über die Teilnahme am sog. Netzwerk der Interessenträger (Stakeholder Network). Dieses wird von der Kommission aus Patienten- und Verbrauchervereinigungen, NGOs aus dem Gesundheitsbereich, Entwicklern und Angehörigen der Gesundheitsberufe aufgebaut. Zu diesem Zweck dürfte in der näheren Zukunft mit der offenen Aufforderung zur Einreichung von Bewerbungen zu rechnen sein. Mit der Bewerbung sind Transparenzpflichten bezüglich finanzieller Quellen und sonstiger Interessen in der Branche der Entwicklung von Gesundheitstechnologien verbunden. Die Mitglieder des Netzwerks einschließlich der Transparenzerklärungen werden über eine IT-Plattform öffentlich zugänglich gemacht.
Wie intensiv sich der Austausch des Netzwerks mit der Koordinierungsgruppe und ihrer Untergruppen in der Praxis gestalten wird, bleibt abzuwarten. Regelmäßig vorgesehen ist ein Treffen lediglich „mindestens einmal jährlich“ (Art. 29 Abs. 5). Außerdem konsultiert die Koordinierungsgruppe das Netzwerk bei der Aufstellung des Jahresarbeitsprogramms (Art. 6 Abs. 3 lit. d) und im Rahmen des sog. Horizon Scanning (Art. 22 Abs. 2 lit. e). Im Übrigen zieht die Koordinierungsgruppe bzw. die Untergruppen das Netzwerk nur nach Bedarf hinzu (Art. 29 Abs. 1). Auch die Teilnahme von Mitgliedern des Netzwerks als Beobachter bei den Sitzungen steht im Ermessen der Koordinierungsgruppe (Art. 29 Abs. 6).
III. Bedeutung der HTA-Verordnung für die Erstattung von Medizinprodukten nach dem SGB V
Bei der Entscheidung über den Zugang und die Kostenübernahme im deutschen Gesundheitssystem werden die EU-HTA-Bewertungen an verschiedenen Stellen zu berücksichtigen sein. Zu nennen sind insbesondere:
- Die Erstattung als Hilfsmittel und die Fortschreibung des Hilfsmittelverzeichnisses: Sie richtet sich nach den Vorgaben des §§ 33, 126 ff. SGB V und den Hilfsmittel-Richtlinien des G-BA. Im Hilfsmittelverzeichnis (§ 139 SGB V) sind alle von der Leistungspflicht umfasste Hilfsmittel aufgeführt. Voraussetzung für die Aufnahme kann u.U. der Nachweis des medizinischen Nutzens sein.
- Neue Untersuchungs- und Behandlungsmethoden in der vertragsärztlichen und vertragszahnärztlichen Versorgung (§ 135 Abs. 1 Satz 1 SGB V) und die Erstattung nach einer neuen EBM-Ziffer: Diese dürfen zu Lasten der Krankenkassen nur erbracht werden, wenn der G-BA auf Antrag in Richtlinien nach § 92 Abs. 1 Satz 2 Nr. 5 SGB V Empfehlungen abgegeben hat u.a. über die Anerkennung des diagnostischen und therapeutischen Nutzens der neuen Methode sowie deren medizinische Notwendigkeit und Wirtschaftlichkeit.
- Die Bewertung von Untersuchungs- und Behandlungsmethoden im Krankenhaus (§ 137c Abs. 1 SGB V), die durch den G-BA darauf geprüft werden, ob sie für eine ausreichende, zweckmäßige und wirtschaftliche Versorgung der Versicherten unter Berücksichtigung des allgemein anerkannten Standes der medizinischen Erkenntnisse erforderlich sind.
- Neue Untersuchungs- und Behandlungsmethoden im Krankenhaus mit Medizinprodukten hoher Risikoklasse (§ 137h Abs. 1 SGB V) und ihre gesonderte Vergütung: Wird hinsichtlich einer neuen Untersuchungs- oder Behandlungsmethode, deren technische Anwendung maßgeblich auf dem Einsatz eines Medizinprodukts mit hoher Risikoklasse beruht, erstmalig eine Anfrage nach § 6 Abs. 2 Satz 3 des KHEntgG mit dem Ziel einer gesonderten Vergütung an das InEK gestellt (sog. NUB-Anfrage), hat das anfragende Krankenhaus im Einvernehmen mit dem Hersteller des Medizinprodukts dem G-BA zugleich Informationen über den Stand der wissenschaftlichen Erkenntnisse zu dieser Methode zur Bewertung zu übermitteln.
IV. Ausblick
Die HTA-Verordnung ist ein wichtiger Schritt zu einer einheitlichen Bewertung von innovativen Hochrisikomedizinprodukten in den Krankenversicherungssystemen der Mitgliedstaaten, auch wenn die Entscheidung über die Kostenübernahme letztlich diesen überlassen bleibt.
Der Umstand, dass die gemeinsame klinische Bewertung gegenüber dem Kommissionsentwurf keinen verbindlichen Charakter mehr aufweist, bedeutet nicht, dass das EU-HTA keinen echten Mehrwert schaffen wird. Denn die nationalen HTA-Stellen sind über die Koordinierungsgruppe unmittelbar in die klinische Bewertung auf europäischer Ebene eingebunden. Praktisch wird es im Rahmen der Erstattungsverfahren auf Ebene der Mitgliedstaaten deshalb einigen Argumentationsaufwands bedürfen, eine positive HTA-Bewertung auf EU-Ebene – ggf. auch in Widerspruch zum Vorgehen anderer EU-Mitgliedstaaten – auf nationaler Ebene wieder in Frage zu stellen. So geht auch bereits der G-BA davon aus, dass die gemeinsame klinische Bewertung auf europäischer Ebene in die frühe Bewertungsentscheidung auf Grundlage des SGB V über einen patientenrelevanten Zusatznutzen einbezogen werden muss, selbst wenn die Erstattungsentscheidung formal nicht vorweggenommen wird.
Aus Sicht der Hersteller von Medizinprodukten stellt zudem die mit der MDR verzahnte Beratungsmöglichkeit eine Option dar, zu einem frühen Entwicklungszeitpunkt – ähnlich wie dies bereits im Arzneimittelbereich möglich ist – eine Vorabbewertung auch von noch in Entwicklung befindlichen Produkte zu erhalten.
Auch wenn die Souveränität der Mitgliedstaaten damit formal von der HTA-Bewertung nicht in Frage gestellt wird, ist ein mit der schrittweisen Etablierung der HTA-Verordnung einhergehender Prozess wachsender Akzeptanz des EU-HTA in den Mitgliedstaaten zu erwarten.