
News on the IVDR/MDR: Extension of transition periods under the IVDR / new Eudamed modules now functional / guidance on classification under the MDR
Currently, several changes and other news items regarding medical device law, more precisely the EU Regulations 2017/745 on medical devices (MDR) and 2017/746 on in vitro diagnostic medical devices (IVDR), have been published. An expansion of the transitional provisions for the IVDR is in the legislative process (I.), new EUDAMED modules are now available (II.), the Medical Devices Coordination Group (MDCG) has issued a Guidance on the Classification of Medical Devices (III.) and the Helsinki procedure has been adapted to the MDR and the IVDR (IV.).
I. Commission proposes expansion of transitional provisions for in vitro diagnostic medical devices
On 14 October 2021, the EU Commission published a Proposal for a Regulation amending Regulation (EU) 2017/746 - IVDR. The proposal does not provide for any substantive changes, but rather an extension of the transitional periods during which in vitro diagnostic medical devices placed on the market and certified under the previous rules can continue to be placed on the market and sold. This is to avoid disruptions in the supply of in vitro diagnostic medical devices, such as HIV tests, pregnancy tests or SARS-CoV-2 tests. However, the date of the IVDR’s entry into force, 26 May 2022, will remain unchanged. One of the most important practical implications of the IVDR concerns the involvement of independent conformity assessment bodies (hereinafter “notified bodies"). Currently, only about 8% of all in vitro diagnostic medical devices on the market are subject to such a procedure due to their risk class.
However, according to the classification rules of the IVDR, about 80% of in vitro diagnostic medical devices will have to be certified by notified bodies before being placed on the market. For manufacturers, this means that they will have to apply to a notified body and obtain one or more certificates after completing the relevant conformity assessment procedure before they can place their products on the market. Such a process takes about one year on the average, depending on the respective risk class of the product, after which around six more months are needed to manufacture the products and place them on the market.
The Commission finds it likely that due to the exceptional circumstances in the context of the COVID-19 pandemic, the relevant parties will not be able in a position to ensure the proper implementation and full application of that Regulation from 26 May 2022. In particular, there is a grave shortage of notified body capacity, making it impossible for manufacturers to conduct the legally required conformity assessment procedures in time. It is not clear at present whether or when a sufficient number of notified bodies will be available to conduct conformity assessment procedures under the IVDR. Currently, only six notified bodies are designated under the IVDR and eleven application procedures are ongoing.
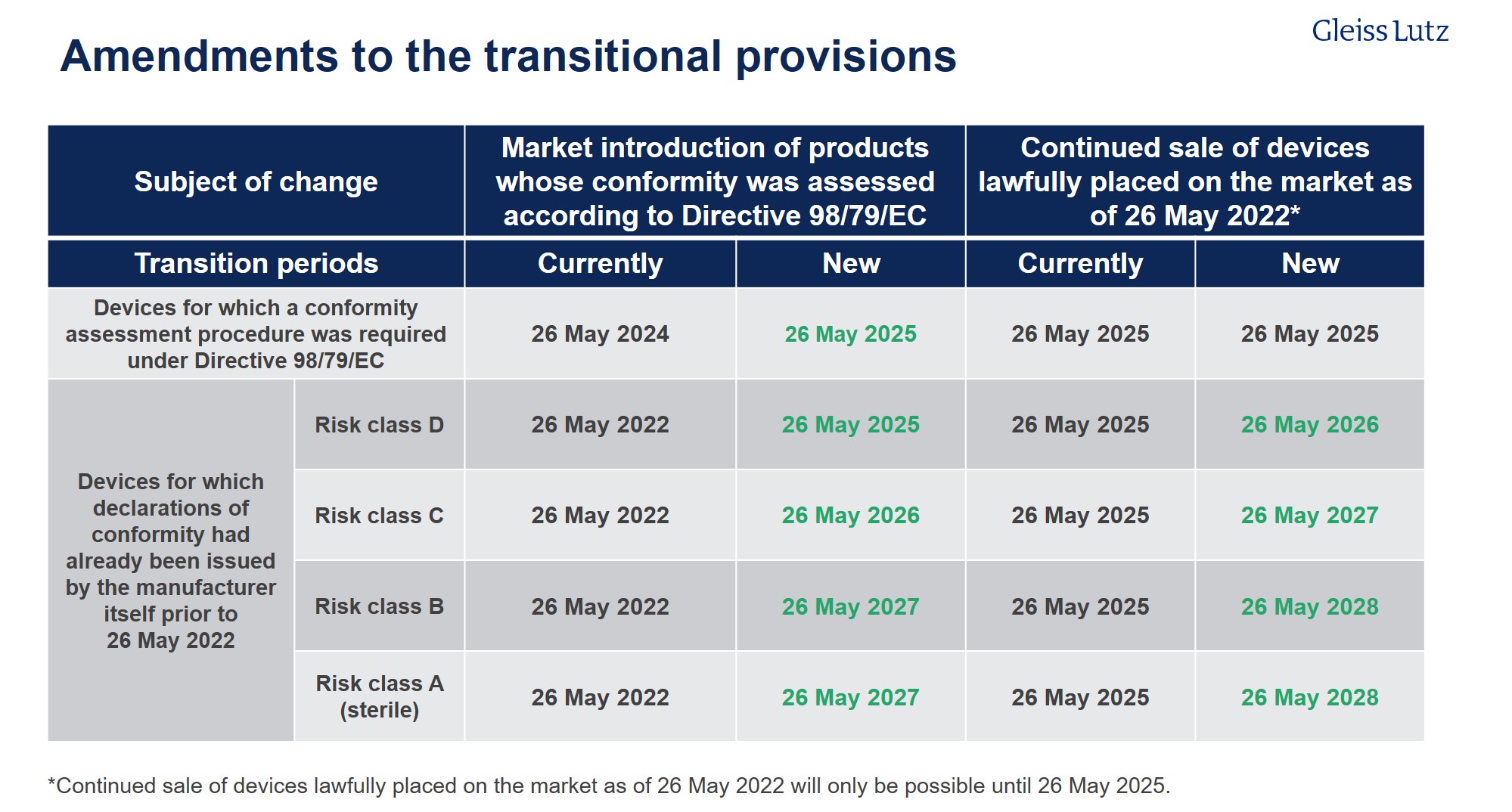
Article 110 IVDR already contains in its current version transitional provisions for those devices for which a conformity assessment procedure was already required under Directive 98/79/EC. These transitional arrangements are to be extended to 27 May 2025. At the same time, transitional periods, graduated according to risk class, will be introduced for those devices for which declarations of conformity had already been issued by the manufacturer itself before 26 May 2022 without the involvement of a notified body but which now must undergo conformity assessment by a notified body for the first time in accordance with Regulation (EU) 2017/746. The higher the risk class, the earlier the in vitro diagnostic medical devices will have to comply with the rules of the IVDR.
Furthermore, the selling-off periods for medical devices placed on the market within the (extended) transitional periods due to Directive 98/79/EC will be extended.
A tabular comparison of the previous transition periods and the extended periods of the Commission proposal including the respective sell-off periods can be found here.
{kind=link}
The Commission is against postponing the date of application of the IVDR as a whole, as it considers the biggest problem to be the limited notified body capacity. This “bottleneck” is to be expanded by spreading the devices that need to undergo a conformity assessment over a longer period of time, allowing for a gradual phase-in of the new Regulation’s requirements, while prioritising high-risk in vitro diagnostic medical devices.
II. New EUDAMED modules on product recognition and registration, notified bodies and certificates
The EUDAMED medical devices database will provide an overview of all medical devices available in the European Union.
The development of EUDAMED is progressing. After the first module, on actor registration, went into operation in December 2020, the second and third of six modules, on unique device identification (UDI) and device registration, as well as on notified bodies and certificates, have been available since the beginning of October 2021.
Data can already be entered there on a voluntary basis. The remaining modules, as well as the functions of the mechanism for scrutiny and the clinical evaluation consultation procedure (CECP) functionalities, will be released as soon as EUDAMED is fully functional.
III. MDCG Guidance on Classification of Medical Devices
Furthermore, the Medical Device Coordination Group (MDCG) has issued MDCG 2021-24, a Guidance on the Classification of Medical Devices. This contains information on the purpose and practical relevance of classification, the implementation of classification and the application of classification rules, as well as a general explanation of the rules and issues that arise.
IV. Updated version of the Helsiniki Procedure
Finally, an updated version of the Helsinki for the MDR and the IVDR was published, the purpose of which is to allow consultation among national competent authorities on borderline and classification issues concerning medical devices or IVDs in comparison with other product groups.